Link to the paper: Molecular Dynamics Simulation of the Stress-Strain Behavior of Polyamide Crystals
Cite this paper: Macromolecules 2021, 54, 18, 8289–8302
Abstract:
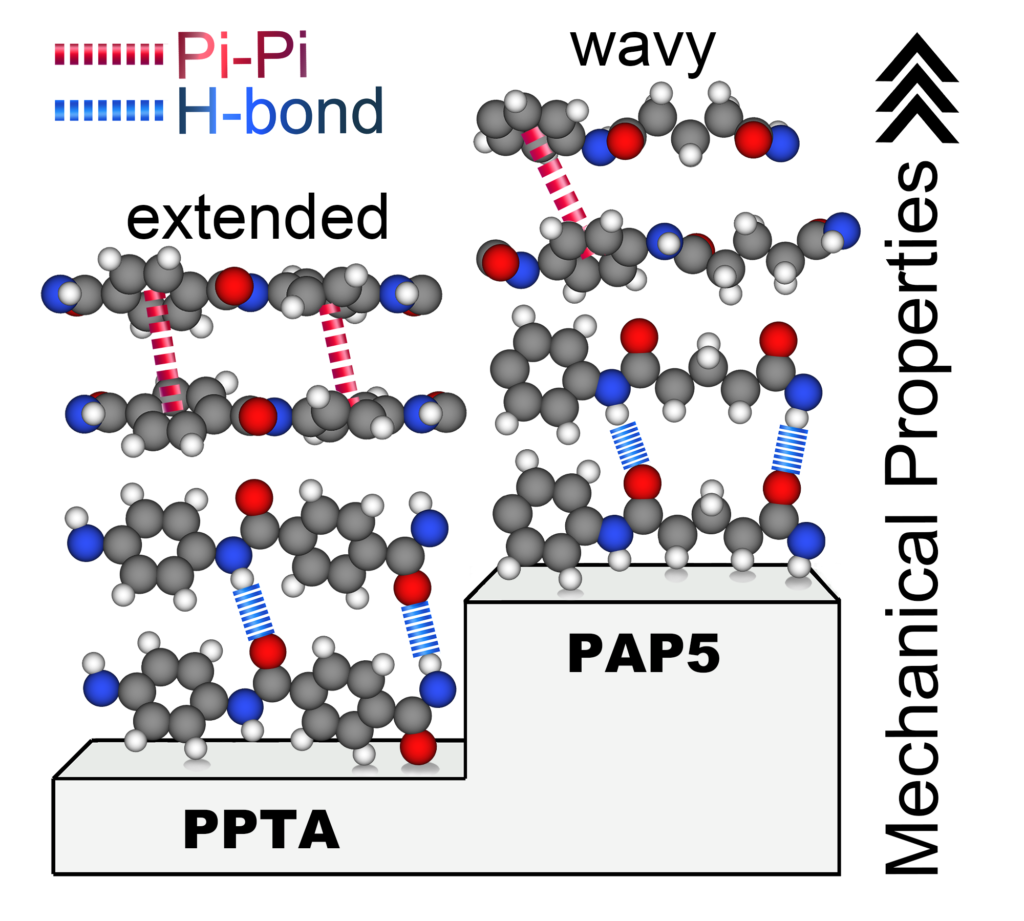
Molecular dynamics simulations were used to model aramid poly(p-phenylene terephthalamide) (PPTA) and a related aromatic–aliphatic polyamide derived from a five-carbon aliphatic diacid (PAP5) with nine different reactive and nonreactive force fields. The force fields were evaluated based on crystal structures as well as intermolecular H-bonding and π-π interactions. An optimum force field was then used to simulate the stress–strain behavior in the chain and transverse-to-chain directions. In the chain direction, PAP5 had higher ultimate stress and failure strain than PPTA; however, the stiffness of PAP5 was lower than that of PPTA at low strain (0–2%), while the reverse was observed at high strain (last 5% before failure). This contrast and the differences in the transverse direction properties were explained by the methylene segments of PAP5 that confer conformational freedom, enabling accommodation of low strain without stretching the covalent bonds. The simulation approach demonstrated here for two polymers with distinct chemistry but similar atomic interactions may be extended to other polyamides.
ToC Graphic: