Abstract:
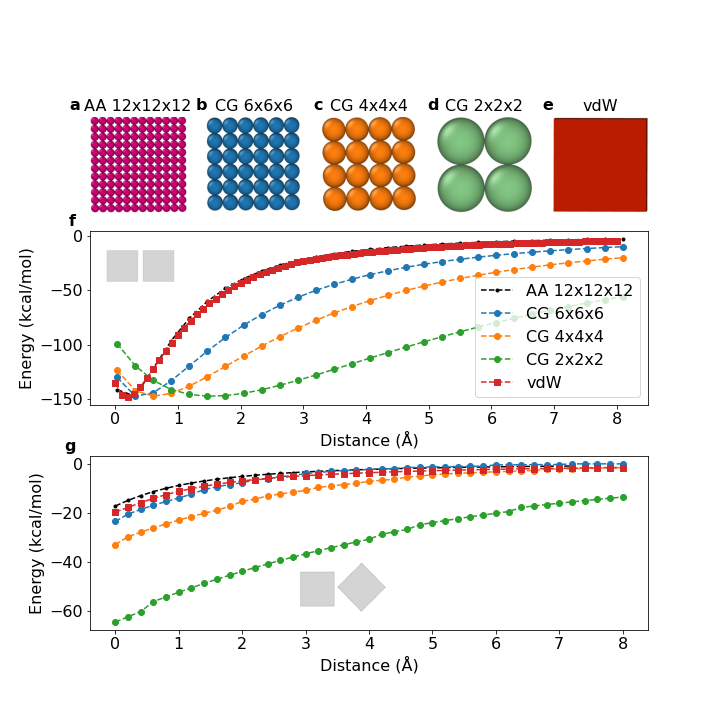
The assemblies of faceted nanoparticles exhibit useful optical, catalytic, and plasmonic properties due to their sharp edges and flat surfaces. Therefore, the development of fast and accurate potential calculation is crucial for investigating their self-assembly behavior. We recently introduced a novel rod-surface discretization approach to derive accurate analytical potentials. These potentials describe the orientation-dependent van der Waals interactions between faceted nanoparticles.
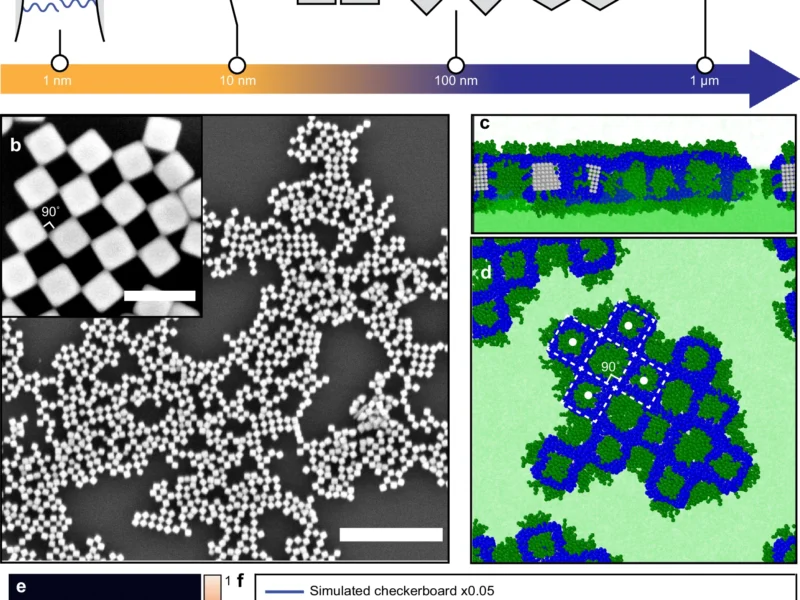
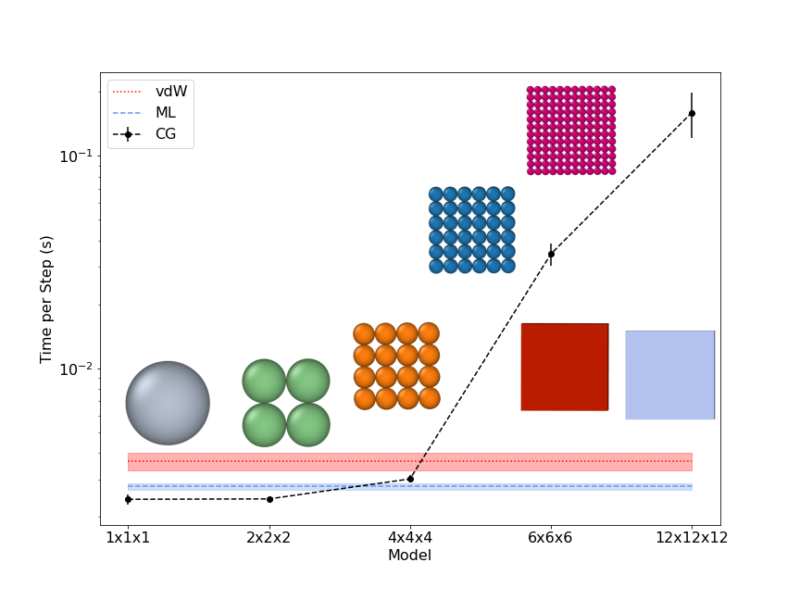
Here, for the first time, we present a Monte Carlo simulation framework that utilizes this analytical vdW potential to rapidly simulate the self-assembly behavior of faceted nanoparticles using nanocubes as an example. We compare its results with atomistic and coarse-grained models. Through the implementation of virtual cluster moves in this framework, we mitigate unphysical energy traps and accurately capture size-dependent diffusive behavior.
Our findings demonstrate the ability of this approach to simulate nanocube assembly dynamics orders of magnitude faster than atomistic models. The morphologies produced closely resemble those obtained from atomistic simulations, whereas coarse-grained models fail to capture the expected self-assembly behavior and morphology.