Listen to the podcast of this paper:
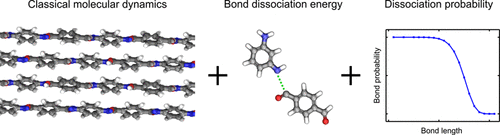
Abstract: Molecular dynamics simulations of the tensile ultimate properties of polymer crystals require the use of empirical potentials that model bond dissociation. However, fully reactive potentials are computationally expensive such that reactive simulations cannot reach the low strain rates of typical experiments. Here, we present a hybrid approach that uses the simplicity of a classical, nonreactive potential, information from bond dissociation energy calculations, and a probabilistic expression that mimics bond breaking. The approach is demonstrated for poly(p-phenylene terephthalamide) and, with one tunable parameter, the calculated tensile ultimate stress matches that obtained using a fully reactive simulation at high strain rates. Then, the hybrid simulations are run at much lower strain rates where the ultimate tensile stress is strain rate-independent and consistent with the expected experimental range.
This is my tenth and last paper during my PhD at UC Merced. It is also the fourth paper from the polyamide project in collaboration with ExxonMobil. It was a fruitful project and I learned and grew mostly from doing this project in my PhD. Thanks everyone!